VASP Bader电荷计算的高级技巧?
创始人
2025-09-05 23:32:45
0次
VASP(Vienna Ab initio Simulation Package)是一种广泛应用于材料科学和化学领域的第一性原理计算软件,用于模拟材料的电子结构和性质。在VASP中进行Bader电荷分析是一种重要的方法,用于分析原子间的电荷转移和电子分布情况。Bader电荷分析基于Richard Bader提出的“原子在分子中”(Atoms in Molecules, AIM)理论,通过电子密度的拓扑结构来划分原子区域,从而计算每个原子的电荷分布。深圳华算科技有限公司将介绍Bader电荷计算的高级技巧。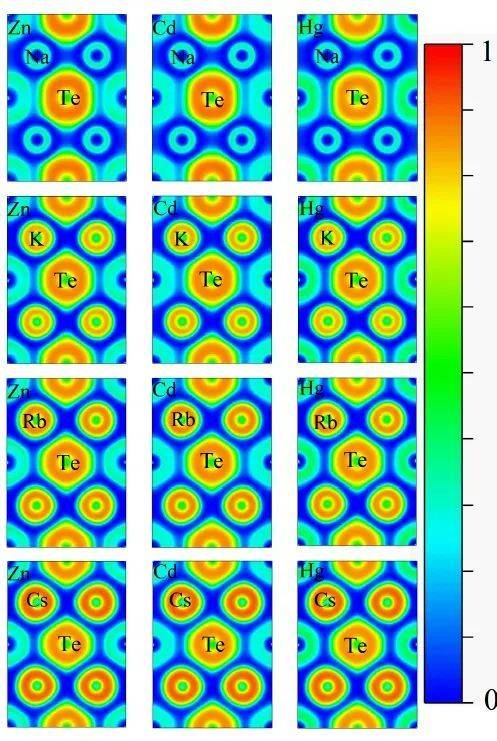
Bader电荷分析的基本原理
Bader电荷分析的核心思想是通过电子密度的零通量面(zero-flux surface)来划分原子区域。在电子密度的拓扑结构中,原子核是电子密度的极大值点,而零通量面是电子密度梯度为零的等值面。 通过这些零通量面,可以将整个电子密度划分为不同的原子区域,从而计算每个原子的电荷分布。这种方法能够更准确地描述原子间的电荷转移和化学键的形成,相比传统的Mulliken电荷分析等方法更为合理。VASP中Bader电荷分析的实现步骤
在VASP中进行Bader电荷分析通常需要以下几个步骤: 结构优化与静态计算 首先,需要对目标结构进行结构优化,以获得稳定的几何构型。优化完成后,进行静态计算,以获得电子结构信息。在静态计算的INCAR文件中,需要设置LAECHG = .TRUE.和LCHARG = .TRUE.,以生成包含核心电荷和价电子电荷的文件(AECCAR0、AECCAR1、AECCAR2)。 后处理工具的准备 Bader电荷分析需要使用Bader程序和chgsum.pl 脚本。Bader程序用于计算Bader电荷,而chgsum.pl 脚本用于将核心电荷和价电子电荷合并为总电荷密度文件(CHGCAR_sum)。 运行Bader程序 使用Bader程序处理生成的电荷密度文件,生成Bader电荷分析结果文件(ACF.dat、BCF.dat、AVF.dat)。其中,ACF.dat文件包含每个原子的Bader电荷信息,是分析电荷转移的关键。 结果分析与可视化 通过分析ACF.dat文件中的电荷信息,可以了解原子间的电荷转移情况。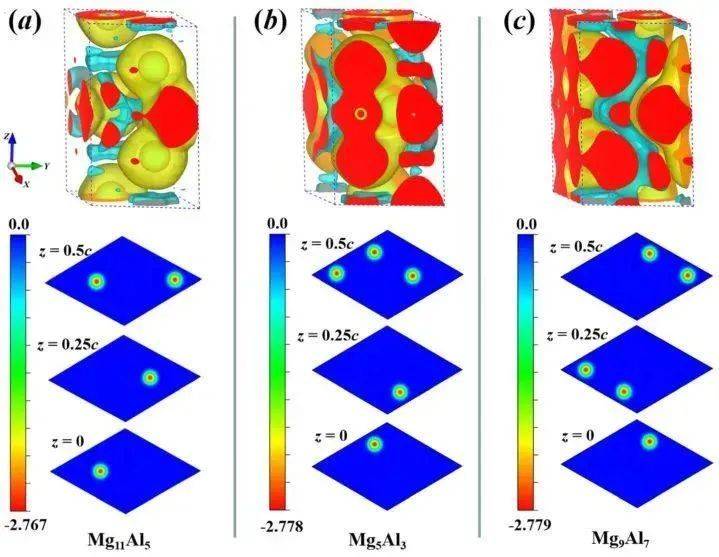
高级计算技巧
结构优化充分优化模型结构,确保原子位置和晶胞参数精确性。在优化过程中,需要设置合适的收敛标准和算法,以确保计算结果的准确性。K点网格密度建议使用Gamma中心K点生成方式,并使用比结构优化更改的K点密度,以提高计算精度。Bader电荷计算在计算Bader电荷时,需要确保chgsum.pl和bader脚本正常安装,并配置好环境变量,确保计算结果的准确性。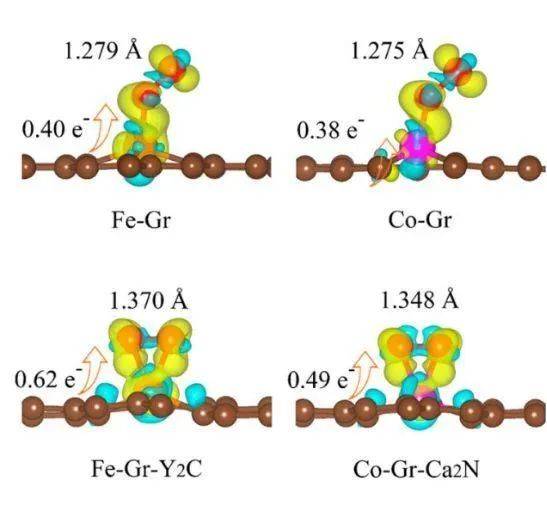
相关内容
热门资讯
西部超导:4月7日融资买入58...
证券之星消息,4月7日,西部超导(688122)融资买入5816.5万元,融资偿还7934.74万元...
博众半导体申请蓝膜芯片剥离装置...
国家知识产权局信息显示,苏州博众半导体有限公司申请一项名为“一种蓝膜芯片剥离装置及芯片生产设备”的专...
功率半导体的风向变了:BDS五...
BDS(双向开关)自诞生之初,就肩负着颠覆功率半导体的使命,它使矩阵变换器 (Matrix Conv...
CPO、半导体等算力硬件股集体...
CPO、 半导体等 算力硬件股集体高开, 铭普光磁、 东山精密涨停, 长光华芯、 华丰科技、 德科立...
半导体掀全产业链涨价潮,科创芯...
今日大科技方向集体上涨,盘中存储芯片、消费电子、汽车芯片、光刻机、AI芯片等细分概念涨幅居前,截至发...
爱思开海力士申请半导体装置和制...
国家知识产权局信息显示,爱思开海力士有限公司申请一项名为“半导体装置和制造该半导体装置的方法”的专利...
A股开盘|沪指涨1.03% 半...
四大股指集体高开,上证指数开盘报3930.25点,涨1.03%,深成指开盘报13717.22点,涨2...
强势反攻!半导体设备ETF华夏...
截至2026年4月8日10:16,科创半导体ETF华夏(588170)上涨4.67%,半导体设备ET...
半导体板块走强
臻镭科技涨超10%,思瑞浦、华虹公司、东微半导、赛微微电、北方华创等涨超5%。 (本文来自第一财经)
地球山申请超低频数字发声芯片专...
国家知识产权局信息显示,地球山(苏州)微电子科技有限公司申请一项名为“一种超低频数字发声芯片”的专利...